Background
Tuberous Sclerosis Complex (TSC)
Tuberous Sclerosis Complex is affecting 1 in every 6,000-10,000
individuals. It is a tumor suppressor syndrome with benign tumors
in multiple organs including the brain, skin, kidney, retina, and
heart. TSC is caused by mutations in the tumor suppressor genes
TSC1 and TSC2. There is no strong genotype/phenotype
correlation, although TSC2-associated disease seems to be more severe.
The severity of the symptoms is variable, with some patients mildly
affected and others with severe mental and developmental delay.
Treatment is symptomatic. The identification of mTOR as downstream
target of TSC1 and TSC2, led to the first clinical trials of the
mTOR inhibitor rapamycin as a treatment option for TSC.
Lymphangioleiomyomatosis (LAM)
LAM is a rare disease affecting exclusively women. It is caused
by proliferation of smooth muscle cells in the lungs. Patients present
with dyspnea, become oxygen dependent and often have multiple pneumothoraces.
End-stage LAM patients often undergo lung transplantation. LAM is
also caused by mutations in TSC1 and TSC2.
The TSC1 and TSC2 tumor suppressor genes
The tumor suppressor genes TSC1
and TSC2 are mutated in
Tuberous Sclerosis Complex and pulmonary Lymphangioleiomyomatosis.
They encode for two proteins, named hamartin and tuberin, respectively.
Tuberin (200 kDa) contains a GTPase-activating protein domain at
its COOH-terminus. Hamartin (130 kDa) contains a COOH-terminal RhoA
activation domain. Hamartin and tuberin form heterodimers, co-localize
and co-immunoprecipitate.
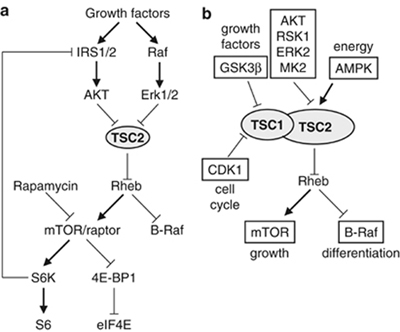
Tuberin negatively regulates the small GTPase Rheb (Figure 1, reviewed
in Astrinidis
and Henske 2005 Oncogene). Upon growth factor stimulation tuberin
is subjected to inhibitory phosphorylation by multiple kinases,
including AKT/PKB, ERK1/2 and MK2, leading to Rheb activation and
increase in the activity of the mammalian target of rapamycin (mTOR)
which regulates mRNA translation, ribosome biogenesis, cell growth,
authophagy, angiogenesis, and apoptosis. Additionally, Rheb negatively
regulates B-Raf kinase which participates in differentiation. Upon
energy starvation and hypoxia, tuberin is positively regulated by
AMPK. Therefore, the hamartin/tuberin complex plays a central role
in integrating signals from different extracellular stimuli.
|
|
Figure 1. (a) Regulation of TSC2/Rheb/mTOR by growth factors.
(b) Integration of growth factor, energy, and cell cycle signals
by phosphorylation-dependent regulation of the hamartin/tuberin
complex (from Astrinidis
and Henske 2005 Oncogene).
Hamartin is phosphorylated during mitosis
Previously we showed that hamartin is phosphorylated by the
CDK1/cyclin B1 complex during the G2/M transition of the cell cycle.
This phosphorylation event negatively regulates the activity
of the hamartin/tuberin complex towards mTOR (Astrinidis
et al. 2003 J. Biol. Chem.). Recently we found that the
hamartin/tuberin complex interacts with the mitotic kinase PLK1.
This interaction is mediated by hamartin residue T310 (Astrinidis
et al. 2006 Hum. Mol. Genet.). We are currently investigating
the role of the hamartin-PLK1 interaction in mitotic progression
and cytokinesis, through the regulation of mTOR and RhoA.
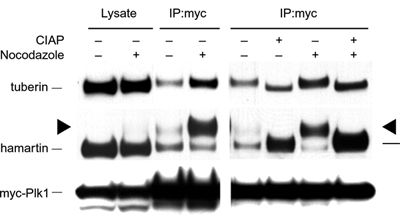
Figure 2. Immunoblotting of control or nocodazole arrested
HEK293 cell lysates, and PLK1 immunocomplexes without or with calf
intestinal alkaline phosphatase (CIAP). Hamartin co-immunoprecipitates
with PLK1. The hamartin present in the PLK1 immunocomplexes is highly
phosphorylated. (Astrinidis
et al. 2006 Hum. Mol. Genet.)
mTOR activation causes centrosome amplification
We found that hamartin localizes to the centrosomes (Figure
3), and that loss of hamartin in cells leads to increased centrosome
number (Figure 4). Pre-treatment of hamartin-deficient cells with
the mTOR inhibitor rapamycin rescues the increased centrosome phenotype (Astrinidis
et al. 2006 Hum. Mol. Genet.). Abnormal centrosome amplification
is observed in several forms of cancers and is directly linked to
instability of the genome. We are currently trying to identify the
molecular pathway leading to centrosome amplification upon TSC1/TSC2
loss and mTOR hyperactivation, and the consequences of TSC1/TSC2
loss in genomic stability.
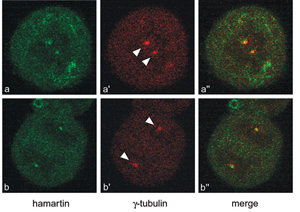
Figure 3. Confocal immunofluorescence images of HeLa cells
showing co-localization of hamartin (green) with the centrosomal
marker gamma-tubulin (red). (Astrinidis
et al. 2006 Hum. Mol. Genet.)
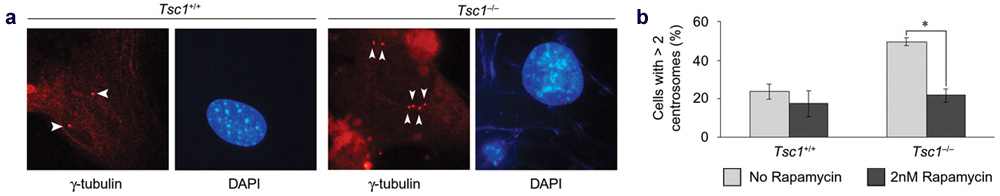
Figure 4. (a) Immunofluorescence micrographs of Tsc1+/+
and Tsc1-/- mouse embryonic
fibroblasts (MEFs) stained with anti-gamma-tubulin (arrowheads).
The Tsc1-/- MEFs have increased
number of centrosomes. (b) Pre-treatment of Tsc1-/-
MEFs with 2nM rapamycin for 24 hours rescues the supernumerary centrosome
phenotype. Asterisk indicates p<0.05. (Astrinidis
et al. 2006 Hum. Mol. Genet.)
mTOR activation increases ploidy
Abnormal centrosome amplification is observed in several forms
of cancers and is directly linked to instability of the genome.
Hamartin-deficient cells have increased DNA content, rescued by
the mTOR inhibitor rapamycin (Figure 5). We are currently trying
to identify the molecular pathway leading to centrosome amplification
upon TSC1/TSC2 loss and mTOR hyperactivation, and the consequences
of TSC1/TSC2 loss in genomic stability.
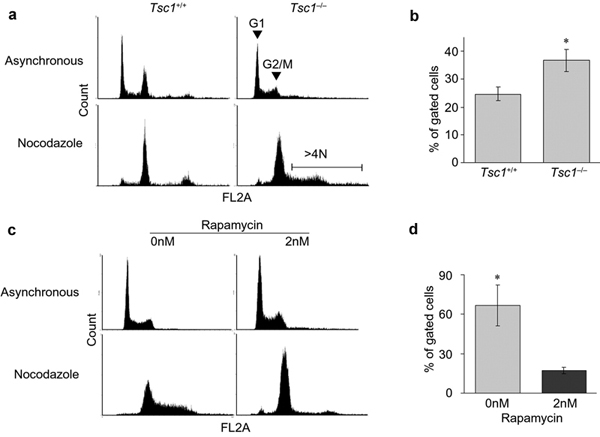
Figure 5. (a) Fluorescence Activated Cell Sorting (FACS)
DNA (FL2) profiles of Tsc1+/+ and Tsc1-/- MEFs after treatment with
vehicle control (asynchronous) or nocodazole. (b) Treatment of Tsc1-/-
MEFs with nocodazole increases the fraction of >4N DNA content
cells. (c, d) Pre-treatment of Tsc1-/- MEFs with 2nM rapamycin rescues
the increased DNA content phenotype. (Astrinidis
et al. 2006 Hum. Mol. Genet.)
Funding and Awareness
Our research is currently funded by the Department
of Defense and Tuberous
Sclerosis Alliance.
Funding for TSC and LAM research is through the NIH,
and specialized programs like the Department of Defense Congressionally
Directed Medical Research Programs. The Tuberous
Sclerosis Alliance, The
LAM Foundation and the LAM
Treatment Alliance provide grants for basic, translational and
clinical research, and have vigorous programs for raising awareness
for the diseases.
|